A portable ultrasound system of the kind that has to clear the regulation before it can carry a CE mark in Europe.
The EU Medical Device Regulation, known as MDR 2017/745, is the law a handheld ultrasound probe has to satisfy before it can carry a CE mark and be sold across the European market. It is not a single test but a whole regime: it decides what class the device falls into, what evidence the maker has to assemble, who has to check that evidence, and what the maker owes its patients after the sale. It replaced an older and looser directive, and the move was deliberately toward more scrutiny, more clinical proof, and more accountability that follows the device through its whole life rather than ending at launch. For a maker outside Europe selling a wireless probe into the continent, the regulation is the gate, and a CE mark on the box is the claim that the gate was passed.
The mark is small, and the file standing behind it is enormous.
Classification sets the burden
The regulation does not treat every device alike. It sorts them by the risk they carry, and the class a probe lands in decides how heavy the path to market will be.
A diagnostic ultrasound probe is an active device intended for diagnosis, and it falls into a middle class rather than the lightest one, which means a maker cannot simply declare conformity on its own and ship. The classification turns on what the device does and how it touches the patient, since a tool whose readings guide a clinical decision carries more consequence than a bandage or a pair of gloves, and the regulation scales its demands to that consequence. Landing in the middle class pulls in a Notified Body, an independent organization that audits the maker’s quality system and reviews the technical documentation before the mark can be applied, so the conformity is checked by an outside party rather than asserted by the maker alone. That single fact separates a serious device from a toy, since the lowest class can self-certify and the middle class cannot, and a buyer who sees a Notified Body number beside the CE mark is seeing evidence that someone independent looked at the file. The classification also shapes how much clinical evidence the maker must bring, how closely the quality system is examined, and how often the device is re-audited once it is on the market, so the class is less a label than the setting that determines the weight of everything that follows.
The class is chosen by what the device does to a patient, and the weight of everything else scales from that one decision.
What the technical file has to prove
At the centre of the regulation sits the technical documentation, the body of evidence that the device is safe and performs as claimed. It is organized around a set of general safety and performance requirements that every device has to meet to the extent they apply.
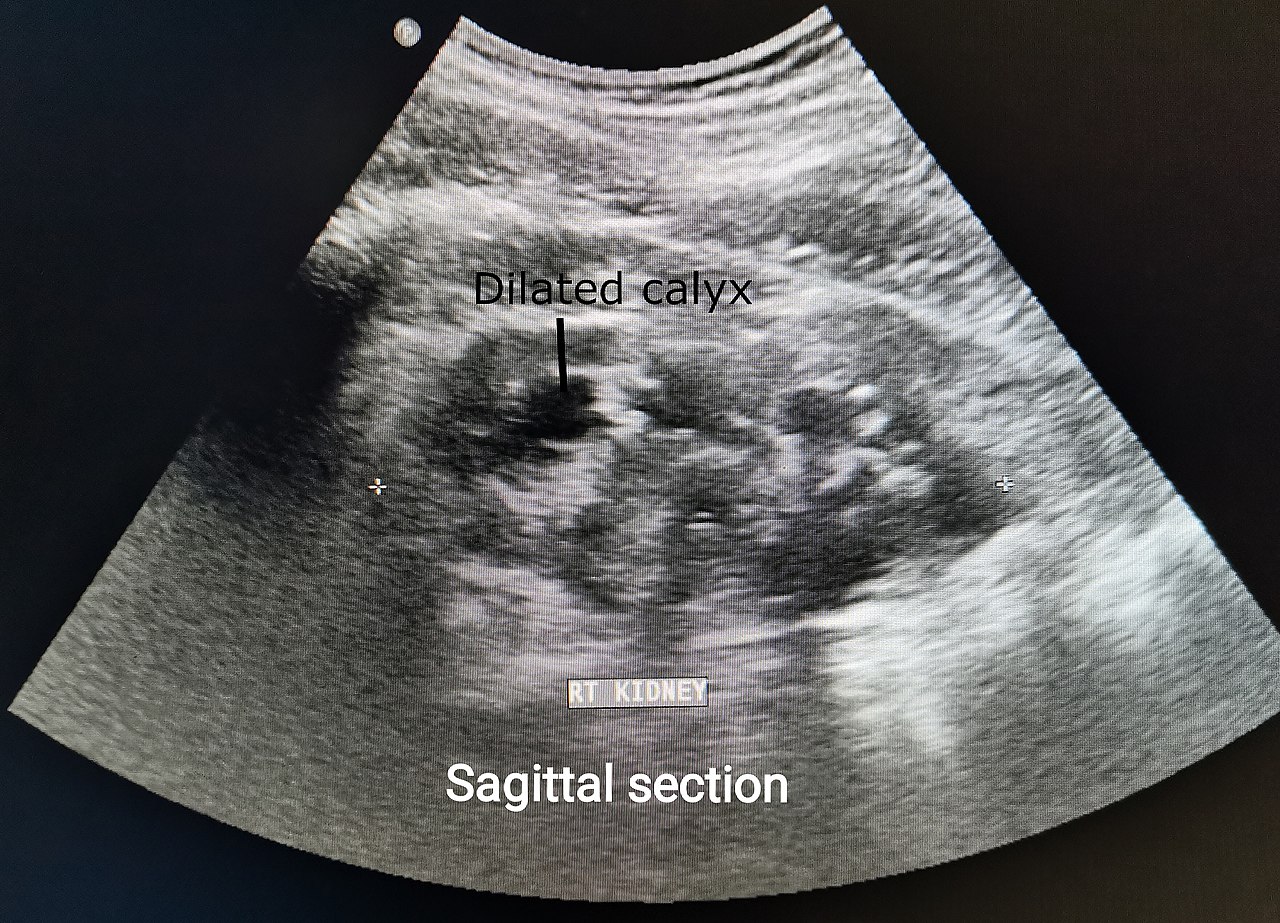
A clinical scan; the regulation asks for clinical evidence that the device performs as claimed, not assertions.
Those requirements are broad by design, covering the physical and chemical safety of the device, its electrical and mechanical soundness, the accuracy of what it measures, the information it gives the user, and the management of every risk it carries. A maker does not satisfy them with assertions; it satisfies them with evidence, and the readiest evidence is conformance to the harmonized standards the regulation recognizes. When a probe meets the particular safety standard for diagnostic ultrasound, the acoustic field measurement standard, the electromagnetic compatibility standard, and the rest, each conformity becomes a building block in the file, and meeting a harmonized standard grants a presumption that the matching requirement has been satisfied. The technical file ties each requirement to the evidence that answers it, the standard met, the test passed, the analysis done, so a reviewer can trace any claim back to its proof. A file that asserts safety without that traceable backing is a file that has not been built, and a Notified Body reading it looks first for the thread that runs from each requirement to its evidence. The clinical evaluation sits alongside, the argument from clinical data that the device does what it claims for patients, and under the regulation that argument has to be more than a citation of similar products waved at the reviewer.
Every requirement in the file points to the evidence that answers it, or the file is not finished.
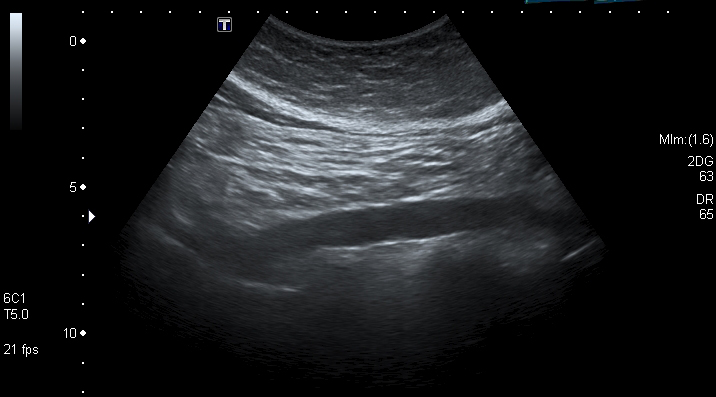
An abdominal scan; the technical file ties each safety requirement to the evidence that answers it.
The duties that outlast the sale
The regulation does not let a maker walk away once the mark is on the box. A large part of it concerns what the maker owes after the device is in the field, the obligations that make the system more than a one-time gate.
A maker has to run a post-market surveillance system that gathers what happens to the device in real use, and for a device of this class that system feeds a post-market clinical follow-up that keeps collecting evidence rather than freezing it at approval. Serious incidents have to be reported to the authorities within fixed timelines, and a pattern of failures can trigger corrective action that the regulator tracks, so a problem found in the field is not a private matter between maker and customer. The device, the maker, and the authorized representative all have to be registered in the European database, and each device carries a unique identifier that lets a specific unit be traced and recalled, which turns an abstract obligation into a concrete chain a regulator can follow. A maker outside Europe has to appoint an authorized representative inside the union who shares legal responsibility, and a designated person responsible for regulatory compliance has to stand behind the file, so accountability has a name and an address rather than dissolving across an ocean. The representative is not a postbox, since the regulation gives that party real duties and real liability, and a buyer who can name the European representative of a foreign maker has found a thread that leads back to someone the law can reach. These after-sale duties are where the regulation bites hardest and where a thin maker stands exposed, since the paperwork to enter the market can be assembled once, but the obligation to watch the device for years is a standing commitment a careless maker quietly fails.
Harmonized standards, and the presumption they buy
The regulation would be unworkable if every maker had to argue each safety requirement from first principles. The harmonized standards are the shortcut the system builds in, and understanding them explains how the whole file holds together.
A harmonized standard is one the union has formally recognized as a way to meet a particular requirement, and conformance to it grants a presumption that the requirement is satisfied, shifting the burden from arguing to demonstrating. For an ultrasound probe the relevant standards are the familiar ones, the particular safety standard that caps acoustic output and governs the on-screen indices, the field measurement standard beneath it, the electromagnetic compatibility standard, the electrical safety standard, and the risk management and biocompatibility standards that sit across the whole device. A maker that meets the recognized version of each can point to that conformance in the file and lean on the presumption rather than reconstructing the safety argument from scratch, which is the efficiency the system is designed to give. The catch is that the presumption holds only for the version of the standard the union has listed, and a maker citing a superseded edition has a file that no longer buys what it claims, so keeping the references current is part of keeping the file alive. A Notified Body reading the file checks that the standards cited are the listed versions and that each requirement they cover is met by them in substance, since a citation is a promise that the standard was met in full rather than merely named. The harmonized standards are where this regulation and all the narrower ones meet, the place a stack of conformities turns into a single coherent claim of safety.
Meet the listed standard and the requirement is presumed met; cite the wrong edition and the presumption evaporates.
What it means for a non-European handheld maker
A wireless ultrasound probe built outside Europe meets this regulation as a foreign entrant, and the regulation is built to make that entry accountable rather than easy.
The maker needs an authorized representative established in the union, a Notified Body engaged to audit and review, a quality system that satisfies the recognized management standard, and a technical file that ties every requirement to its evidence. None of that can be improvised at the border, since the Notified Body audit and the clinical evaluation take time and cannot be bought as a certificate the week before a trade show. A maker that has done the work carries a CE mark with a Notified Body number behind it, a Declaration of Conformity that names the regulation and the standards met, and a representative a buyer can contact inside Europe. A maker that has not done the work offers a CE mark with no number, a self-declaration that quietly applies to the wrong class, or a sticker that means nothing at all, and a buyer who knows the regulation reads those signs accurately. There is a notorious trap in the mark itself, since a similar-looking symbol meaning China Export has been used to blur the line, and the genuine European mark carries proportions and, for this device class, a Notified Body number that the imitation lacks. A buyer who treats the letters alone as proof has read the costume rather than the credential, and the regulation gives that buyer a better test in the declaration and the number behind the mark. The handheld market draws entrants who treat the mark as decoration, and the regulation exists so that a buyer can tell the decoration from the document, provided the buyer knows where to look.
A CE mark with no number behind it is a claim a careful buyer does not take at face value.
Reading a Declaration of Conformity
The document a buyer can ask for first is the Declaration of Conformity, the maker’s signed statement of what the device complies with. A few checks separate a real one from a hollow one.
A sound declaration names the regulation explicitly rather than gesturing at compliance, lists the device by model and identifier, states the class the device falls into, and names the Notified Body with its four-digit number where the class requires one. It lists the harmonized standards the device was built against, so a reader can see that the particular safety standard, the acoustic measurement standard, and the electromagnetic standard are all accounted for rather than skipped. It carries a date, a signature, and the name of the person who stands behind it, since a declaration left unsigned is a claim that has no owner. A declaration that omits the Notified Body number for a device that needs one, or that cites the old directive rather than the current regulation, or that names no standards at all, is telling a careful reader that the work behind it is incomplete. The buyer who reads the declaration against the class the device should hold is checking the one document that ties the whole regime together, and a maker that hands it over without hesitation has usually earned the confidence the gesture invites. A declaration is also cheap to fake in appearance and hard to fake in substance, since the standards it names can be checked against the listed versions and the Notified Body number can be looked up in a public register, and a maker that has invented either has left a thread a diligent buyer can pull. The point of reading it is not to catch a forger but to tell a finished file from an unfinished one, and the difference shows in whether the page can survive a few minutes of checking against the registers behind it.
The declaration is the regulation compressed onto a page, and a hollow page gives a maker away.
About the Author
Julien Mercier
Senior R&D Engineer · Medical Ultrasound Transducer Development
Senior R&D Engineer with an M.S. in Applied Physics and over 15 years of experience in medical ultrasound transducer development, specializing in the design verification and performance testing of high-frequency imaging transducers. Currently leading the development and verification of the company’s next-generation high-frequency linear-array transducer, responsible for imaging performance evaluation and reliability analysis in preclinical testing. Brings extensive hands-on experience in piezoelectric element tuning, beamforming parameter optimization, and system-level performance testing.